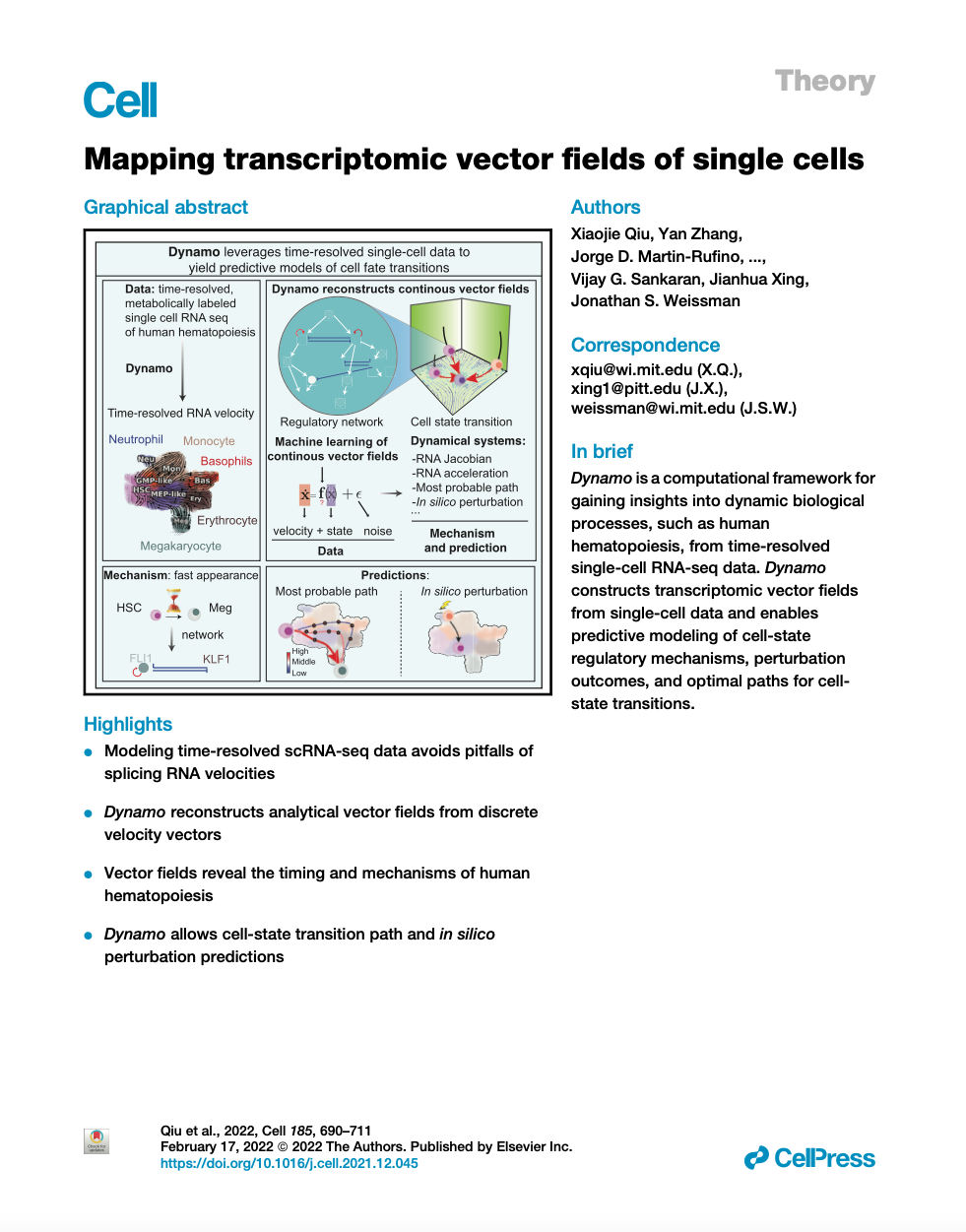
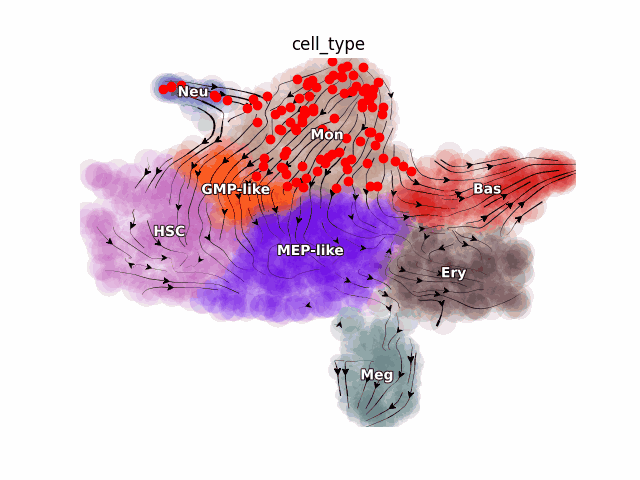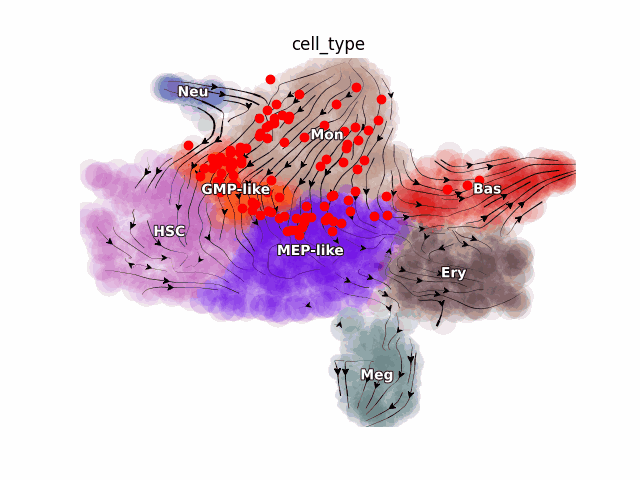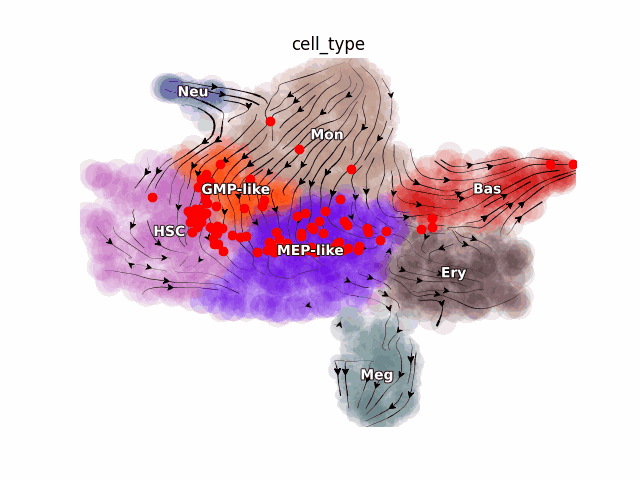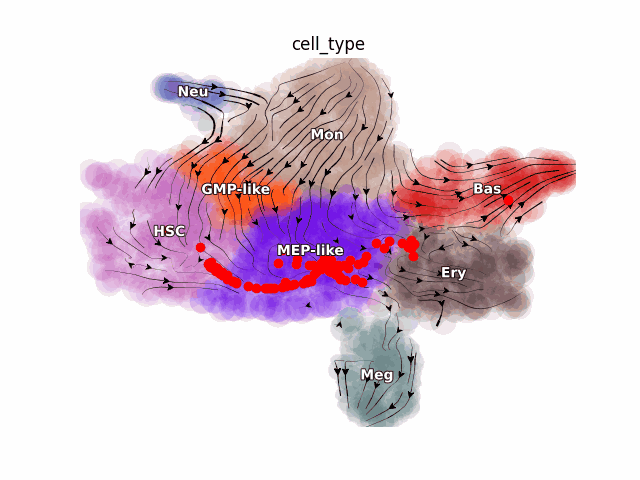
Combine time-resolved single cell genomics with differentiable machine learning approaches
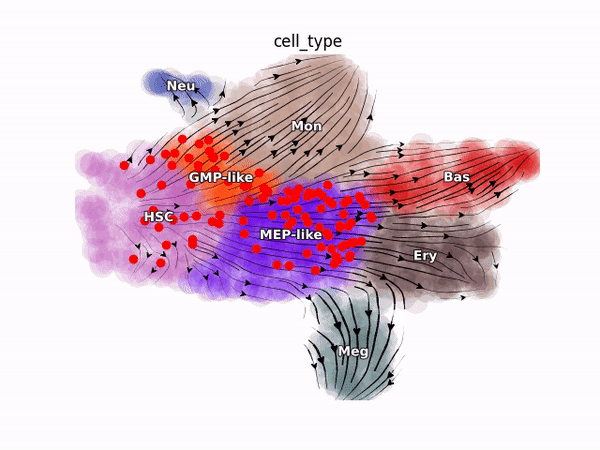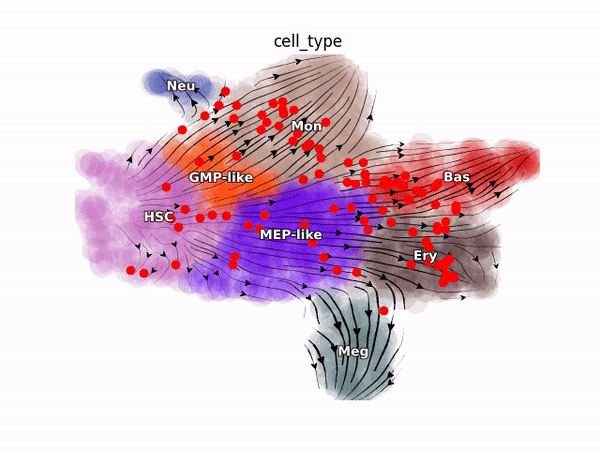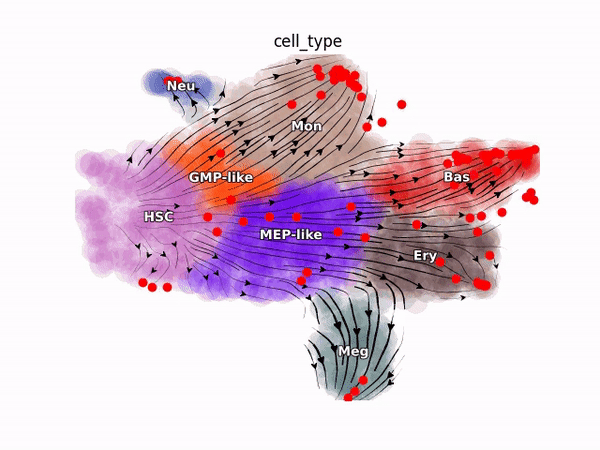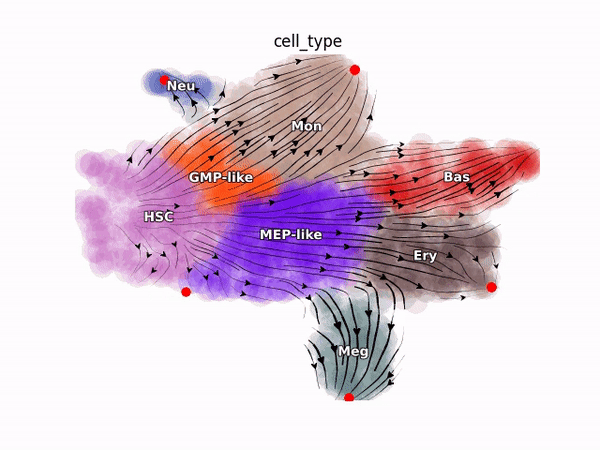
Emergent single-cell genomics have enabled profiling of cell-state transitions with unprecedented scale. However, due to their destructive nature, it is generally infeasible to follow the same cell over time. Advances in single-cell profiling have fueled the development of computational approaches for inferring cellular dynamics from snapshot measurements, such as pseudotime and RNA velocity approaches. Furthermore, exciting developments in metabolic labeling enabled scRNA-seq approaches now enable us to obtain time-resolved transcriptomic kinetics by directly measuring “new” and “old” RNAs in a controllable manner. We recently developed the Dynamo framework that overcomes fundamental limitations of conventional splicing-based RNA velocity analyses to enable accurate velocity estimations for time-resolved scRNA-seq datasets. Furthermore, we go beyond the discrete RNA velocity vectors to a continuous function that can be used to perform higher-order differentials to gain functional biological insights. Dynamo also establishes itself as one of the first tools to make non-trivial predictions of optimal reprogramming paths and in silico perturbation predictions with single cell datasets. We are continuing this line of research in the lab.
Major questions we are actively asking in the lab include:
How to advance Dynamo’s analytical vector field approach to differentiable deep learning frameworks to dissect mechanisms of mammalian cell evolution, differentiation, maintenance, aging, and reprogramming?
How to integrate RNA metabolic labeling with multi-omics experimentally, and then extend the RNA velocity approaches to multi-modal velocity of epigenetics, RNA, and protein?
How to use methods like machine translation to translate single-cell gene expression states to other modalities, such as chromatin accessibility or proteomics, and vice versa?
How to harmonize short-term RNA velocities with long-term cell state transitions measured with CRISPR-Cas9 based or mitochondria mutation-based lineage tracing?
Leverage spatial genomics to model cell-cell interaction and tissue organization in 3D space

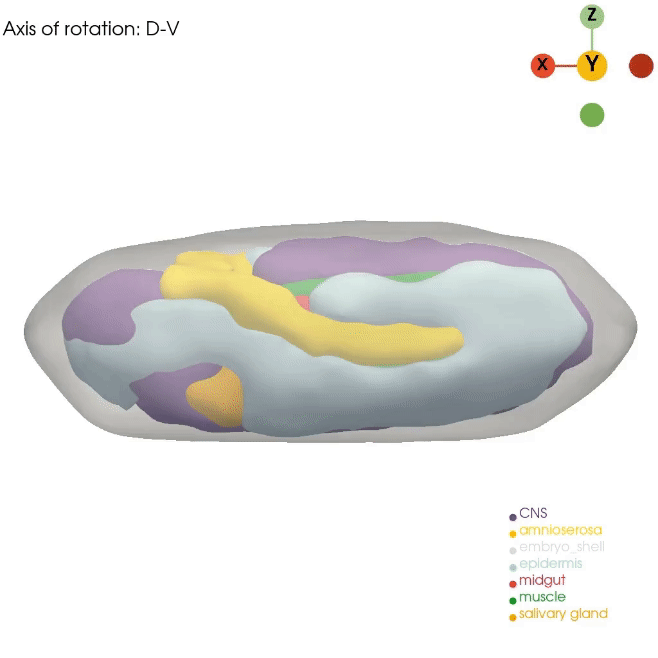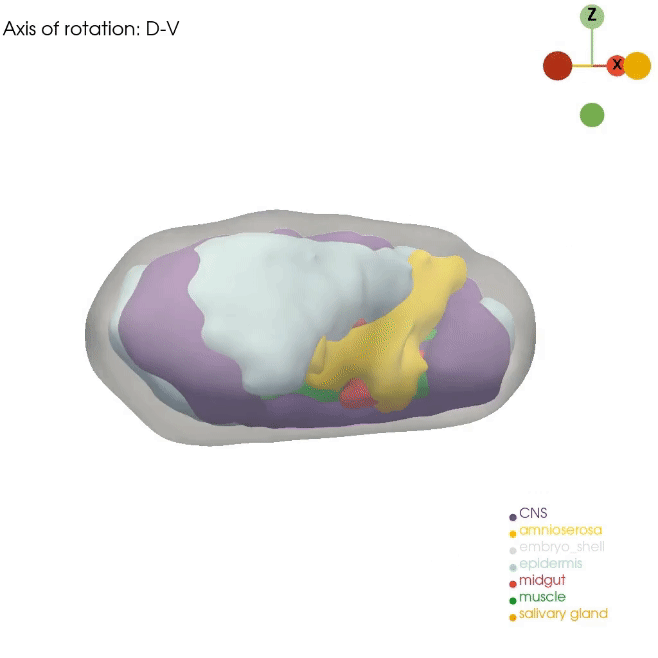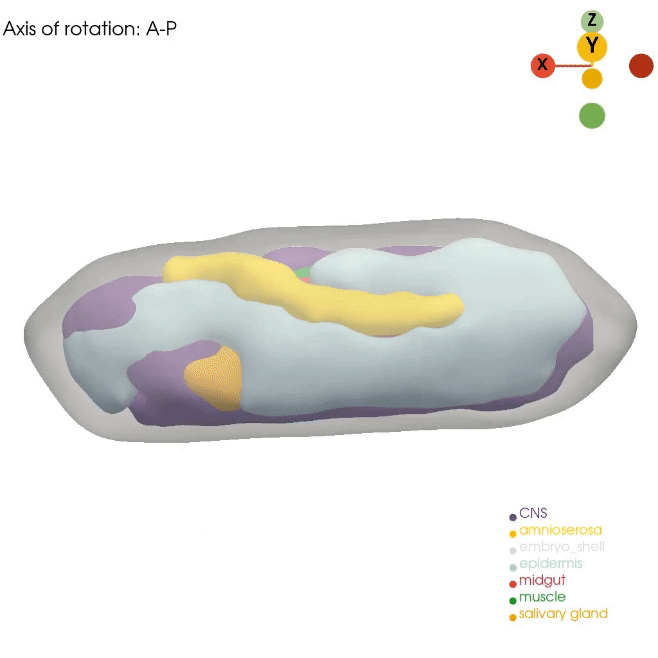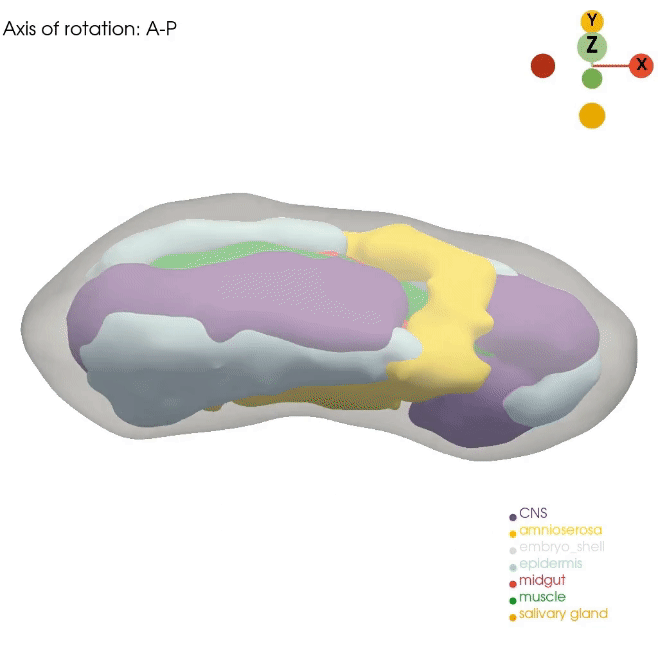
Our Dynamo approach for RNA velocity vector field allows us to gain mechanistic insight into cell state transitions from the temporal axis. However, complex cell fate changes such as embryogenesis are not only temporally controlled but also spatially controlled. Unfortunately, routine single-cell approaches dissociate the cells, resulting in complete loss of spatial information. In order to best model the spatial axis, similar to the Dynamo work, we will apply, optimize, and develop novel technologies, such as Stereo-seq, STARmap and PIXEL-seq, that can generate the ideal spatial data. Although these promising technologies now allow us to profile cell states across subcellular, cellular, tissue, organ, and even whole embryo levels, analytical tools that fully leverage such data remain lacking. We recently developed a general analytical framework, Spateo, to model spatial transcriptomics at a multidimensional scale, ranging from single-cell segmentation, spatial domain clustering and digitization, cell-cell interactions, to whole organ and tissue 3D morphometric analysis. We will also continue this line of research in the lab.
Major questions we are actively asking in the lab include:
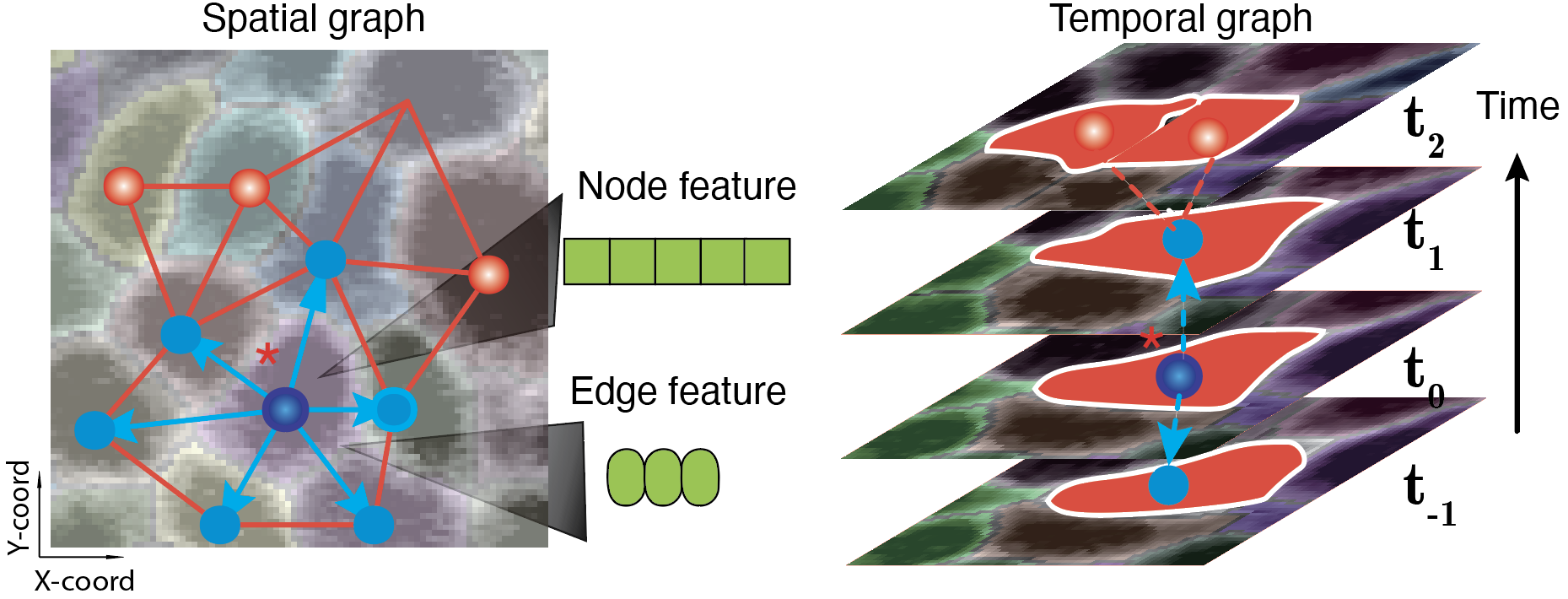
How can we model RNA subcellular distribution and co-localization of image-based spatial transcriptomics using advanced machine learning techniques such as graph neural networks?
How can we properly reconstruct the 3D volumetric structure of whole organs and embryos with serial profiling of spatial transcriptomics using optimal transport and more generalizable and efficient alternatives?
How to enable novel 3D analysis of the reconstructed 3D models with 3D computer vision approaches?
How to model cell-cell communication at cellular, tissue, and organ levels and in 3D space using spatially aware machine learning models?
How can we extend spatial transcriptomics with multi-omics and Perturb-seq to build spatially resolved predictive models for these datasets?
Joint spatiotemporal modeling of single cells
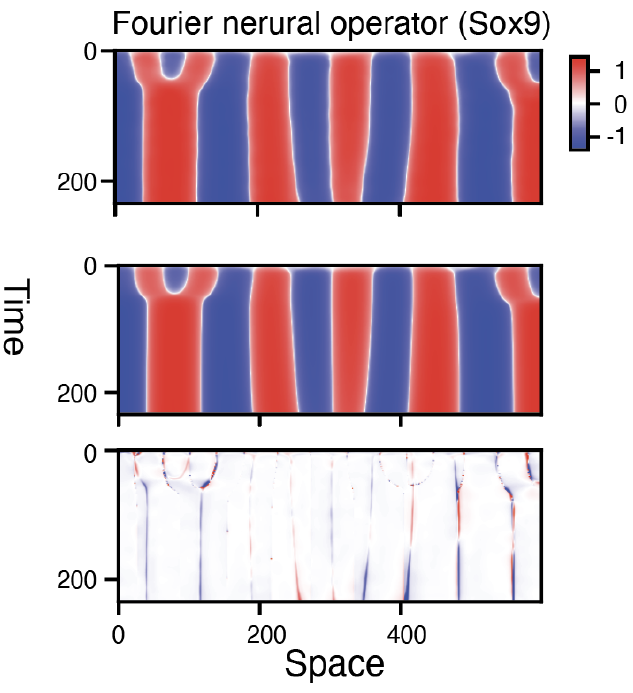
While Dynamo allows us to predict how cells transit in the gene expression space along the temporal axis, the Spateo framework allows us to predict how cells migrate in the physical space. But these two spaces are separate. It is thus critical to build predictive tools to unify both spaces to jointly learn spatiotemporal kinetics. However, spatiotemporal models, such as partial differential equations, are challenging to directly learn from data, especially for high-dimensional biological systems. Fortunately, breakthroughs in Fourier neural operators, diffusion models, and other generative deep learning approaches, which incorporate functional learning, attention and diffusion process, respectively, have begun to show promise for addressing such challenges. We have achieved promising results with these approaches.
In the lab, we are pursuing the following key questions:
How can we integrate RNA metabolic labeling with spatial transcriptomics to obtain real spatiotemporal datasets to power our machine learning model?
How can we leverage graph neural networks and neuralODE, Fourier neural operators and others, to learn spatiotemporal kinetics?
How can we jointly model cell growth, division, apoptosis, and migration with whole organ or embryo datasets in 3D space across multiple time points?
How to in silico perturb genes at a particular time point at a particular spatial location to predict how such perturbations propagate over time and space?
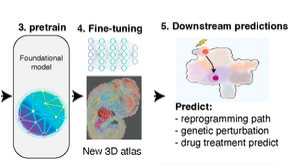
Moving towards cross-species foundational models of single cells
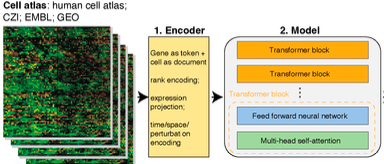
Recent advances in machine learning, particularly with powerful, versatile foundational models like ChatGPT, have transformed various fields. These models use attention based transformer and are trained on extensive datasets, enabling them to perform exceptionally well in areas like image, text, and video processing with limited data. Advances in single-cell and spatial genomics has accumulated massive datasets with tens of millions of single cells that provided necessary data to power up foundational models. While there have been several exciting attempts in building these models, such as Geneformer, scGPT, scFoundation and others, in biology with specific downstream tasks like batch effect removal and gene regulatory network analysis, their predictive capabilities are still lacking. To fulfill these unmet gaps, we have formed a vibrant team to actively investigating these possibilities.
We are pursuing the following key questions:
How to overcome the limitations of treating genes as token and cells as sentences in Geneformer, scGPT, scFoundation and others when building the single cell foundational model?
How to extend existing foundational model to multi-modal datasets using machine translation, tabular learning and others?
How to build cross-species foundational models so that we can unify the gene and cell embeddings across evolutionary scales?
How to utilize spatiotemporal transformers to model the time-resolved and spatially-resolved single cell genomics, and in 3D space?
How to perform non-trivial zero-shot downstream predictions, such as in silico perturbations and others, with the foundational model?
How to model cell growth, division, migration and others to simulate a differentiable in silico model of mamalian embryogenesis and organogenesis?